What is ALS (Amyotrophic Lateral Sclerosis)?
How does muscular atrophy occur?
|
|
|
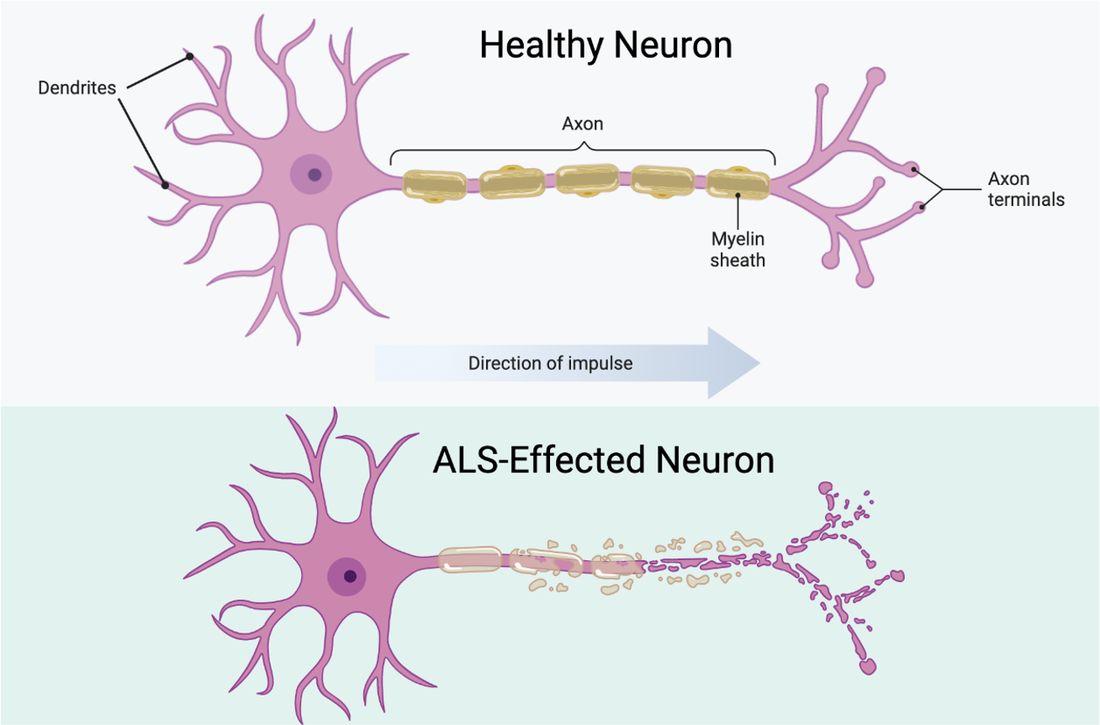
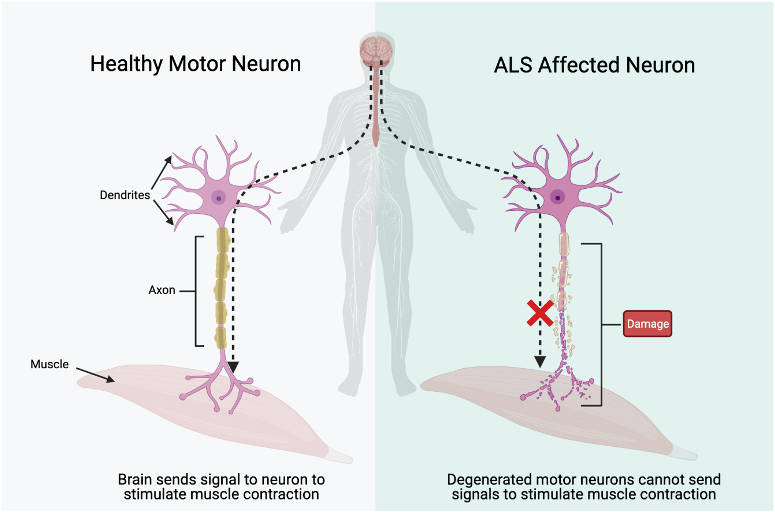
Neurons affected by ALS show a lot of damage to dendrites, the axon, myelin sheath, and axon terminals. Without having these components intact, the impulses from the brain cannot get to the muscle to stimulate a contraction. As a result, these muscles degenerate and breakdown over time due to the lack of use. This breakdown is known as muscular atrophy. |
What is tdp-43 and what is its role in ALS?

|
TDP-43, a protein formally known as transactive response DNA binding protein, is protein encoded by the TARDBP gene, and is 414 amino acids in length. Under normal conditions, this protein resides in the nucleus of most cell tissues, and its dominant purpose is to regulate many steps of protein production. TDP-43 plays a role in regulation of transcription, a process in which an RNA code is copied into a single-stranded complement copy of RNA from the DNA genome by a series of protein factors. This single-stranded RNA strand then provides a sequence that encodes an amino acid sequence that will bend and twist to form a functional protein.
|
We established that TDP-43 holds responsibilities within the processes of transcribing DNA into RNA, and then translating RNA into proteins. These processes occur both within the nucleus and outside of the nucleus within the cytoplasm. Special localization properties of TDP-43 allow the protein to exist in both places with stability.
|
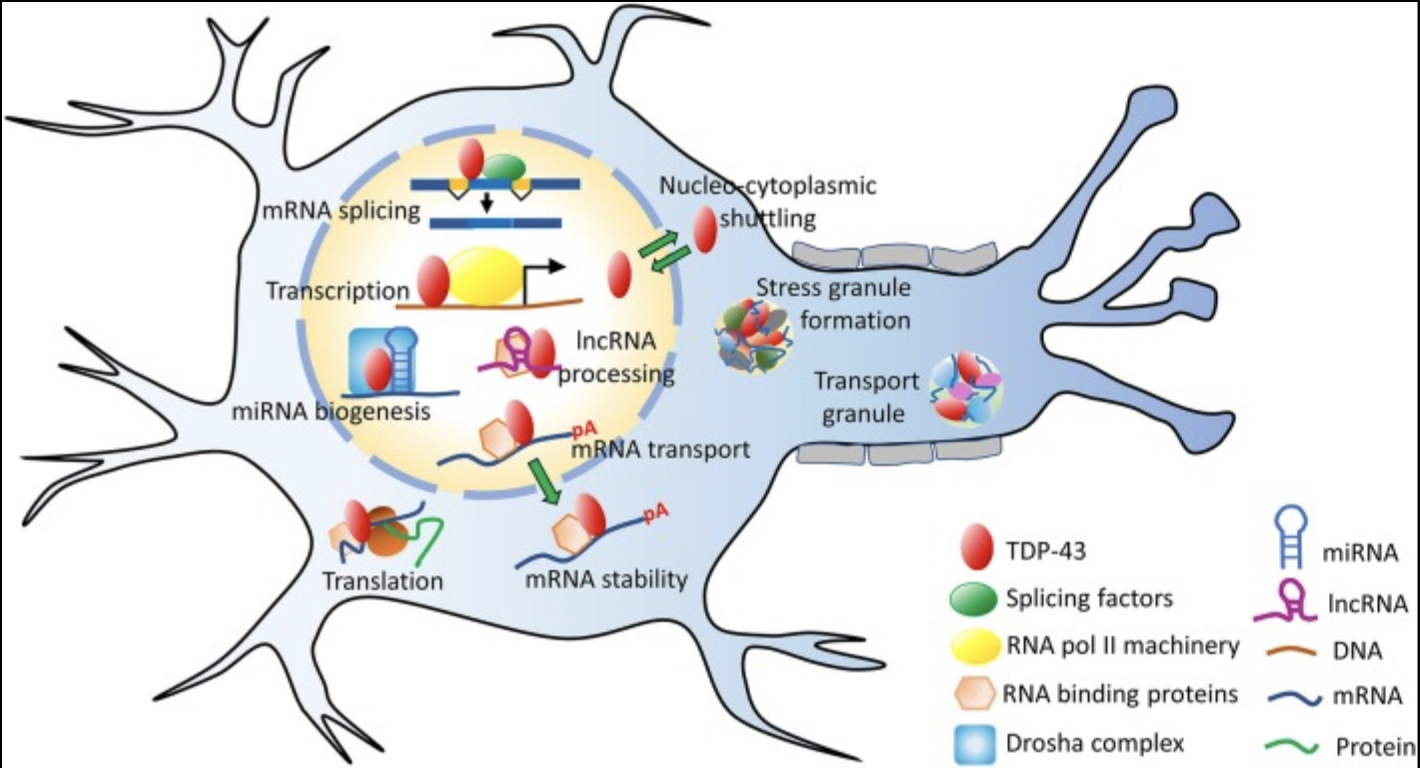
Different regions of a protein perform different roles, meaning mutations within different regions can alter and/or shut off various functions of the protein. The figure to the right depicts the many functions that TDP-43 performs within a neuron. The protein predominantly resides within the nucleus, but does leave the nucleus and enter the cytoplasm to perform certain tasks such as mRNA stabilization and assistance in translation processes in the formation of other proteins.
The localization of TDP-43 is dependent on its hydrophilic properties, or its attraction to water and other ionic charges. In mutated TDP-43, amino acid sections (known as motifs) M1, M3, and M5 contain extensive chains of hydrophobic amino acids. The excess hydrophobic properties cause several TDP-43 proteins to aggregate into a hydrophobic, and insoluble, clusters. The structure of TDP-43 is still unknown, and the discovery of its structure will allow researchers to study just how the mutated protein is twisting in a way that aggregation occurs. |
Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. 2019 Feb 14;12:25. doi: 10.3389/fnmol.2019.00025. PMID: 30837838; PMCID: PMC6382748.
|
With increased mis-localized, unstable, and nonfunctioning TDP-43 buildup, processes like protein formation cannot occur within neuron cells. The lack of ability to fulfill the energy and material needs of the neuron through normal cellular processes causes the degradation of muscular neurons. This degradation is ultimately what causes the lack of ability to transmit the signal from the brain, down the brainstem, and into motor neurons to elicit a contraction. What makes ALS particularly terminal is from eventual breakdown and atrophy of cardiac muscle, causing respiratory failure.
While no cure exists for ALS currently, scientists continue to search for various causes, mutations, and pathways that lead to the ALS phenotype. Studies on the TDP-43 and its specific mechanisms associated with ALS continue to be published, leaving patients, loved ones, and the scientific community hopeful that new targets for therapeutic medicine will be revealed.
While no cure exists for ALS currently, scientists continue to search for various causes, mutations, and pathways that lead to the ALS phenotype. Studies on the TDP-43 and its specific mechanisms associated with ALS continue to be published, leaving patients, loved ones, and the scientific community hopeful that new targets for therapeutic medicine will be revealed.
[1]Gao J, Wang L, Huntley ML, Perry G, Wang X. Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem. 2018 Feb 27:10.1111/jnc.14327. doi: 10.1111/jnc.14327. Epub ahead of print. PMID: 29486049; PMCID: PMC6110993.
[2]Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, Mann DM, Allsop D, Nakagawa M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009 Jan;117(1):55-62. doi: 10.1007/s00401-008-0456-1. Epub 2008 Nov 7. PMID: 18989684.
[3]Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. 2019 Feb 14;12:25. doi: 10.3389/fnmol.2019.00025. PMID: 30837838; PMCID: PMC6382748.
[4]Scotter EL, Chen HJ, Shaw CE. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics. 2015 Apr;12(2):352-63. doi: 10.1007/s13311-015-0338-x. Erratum in: Neurotherapeutics. 2015 Apr;12(2):515-8. PMID: 25652699; PMCID: PMC4404432.
[5]“TARDBP Gene: MedlinePlus Genetics.” MedlinePlus, U.S. National Library of Medicine, 18 Aug. 2020, medlineplus.gov/genetics/gene/tardbp/.
[2]Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, Mann DM, Allsop D, Nakagawa M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009 Jan;117(1):55-62. doi: 10.1007/s00401-008-0456-1. Epub 2008 Nov 7. PMID: 18989684.
[3]Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. 2019 Feb 14;12:25. doi: 10.3389/fnmol.2019.00025. PMID: 30837838; PMCID: PMC6382748.
[4]Scotter EL, Chen HJ, Shaw CE. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics. 2015 Apr;12(2):352-63. doi: 10.1007/s13311-015-0338-x. Erratum in: Neurotherapeutics. 2015 Apr;12(2):515-8. PMID: 25652699; PMCID: PMC4404432.
[5]“TARDBP Gene: MedlinePlus Genetics.” MedlinePlus, U.S. National Library of Medicine, 18 Aug. 2020, medlineplus.gov/genetics/gene/tardbp/.
|
Catherine Eldridge
B.S. Genetics and Genomics '21 Certificate Environmental Studies Certificate Global Health [email protected] 269-870-6505 |
HOME |
CONTACT |

